
Publications
Ben’s full publication list is available at his Google Scholar profile. Selected publications and abstracts are highlighted below.
Gardnerella diversity and ecology in pregnancy and preterm birth
mSystems, 2024
The vaginal microbiome has been linked to negative health outcomes including preterm birth. Specific taxa, includingGardnerella spp., have been identified as risk factors for these conditions. Historically, microbiome analysis methods have treated all Gardnerella spp. as one species, but the broad diversity of Gardnerella has become more apparent. We explore the diversity of Gardnerella clades and genomic species in the vaginal microbiome of pregnant women and their associations with microbiome composition and preterm birth. Relative abundance of Gardnerella clades and genomic species and other taxa was quantified in shotgun metagenomic sequencing data from three distinct cohorts of pregnant women. We also assessed the diversity and abundance of Gardnerella variants in 16S rRNA gene amplicon sequencing data from seven previously conducted studies in differing populations. Individual microbiomes often contained multiple Gardnerella variants, and the number of clades was associated with increased microbial load, or the ratio of non-human reads to human reads. Taxon co-occurrence patterns were largely consistent across Gardnerella clades and among cohorts.
preterm birth. Specific taxa, includingGardnerella spp., have been identified as risk factors for these conditions. Historically, microbiome analysis methods have treated all Gardnerella spp. as one species, but the broad diversity of Gardnerella has become more apparent. We explore the diversity of Gardnerella clades and genomic species in the vaginal microbiome of pregnant women and their associations with microbiome composition and preterm birth. Relative abundance of Gardnerella clades and genomic species and other taxa was quantified in shotgun metagenomic sequencing data from three distinct cohorts of pregnant women. We also assessed the diversity and abundance of Gardnerella variants in 16S rRNA gene amplicon sequencing data from seven previously conducted studies in differing populations. Individual microbiomes often contained multiple Gardnerella variants, and the number of clades was associated with increased microbial load, or the ratio of non-human reads to human reads. Taxon co-occurrence patterns were largely consistent across Gardnerella clades and among cohorts.
Meta-analysis reveals the vaginal microbiome is a better predictor of earlier than later preterm birth
BMC Biology, 2023.

Background: High-throughput sequencing measurements of the vaginal microbiome have yielded intriguing potentialrelationships between the vaginal microbiome and preterm birth (PTB; live birth prior to 37 weeks of gestation). However, results across studies have been inconsistent.
Results: Here, we perform an integrated analysis of previously published datasets from 12 cohorts of pregnant women whose vaginal microbiomes were measured by 16S rRNA gene sequencing. Of 2039 women included in our analysis, 586 went on to deliver prematurely. Substantial variation between these datasets existed in their definition of preterm birth, characteristics of the study populations, and sequencing methodology.
Implications of taxonomic bias for microbial differential-abundance analysis
Biorxiv, 2022.

Differential-abundance (DA) analyses enable microbiome researchers to assess how microbial species vary in relative orabsolute abundance with specific host or environmental conditions, such as health status or pH. These analyses typically use sequencing-based community measurements that are taxonomically biased to measure some species more efficiently than others. Understanding the effects that taxonomic bias has on the results of a DA analysis is essential for achieving reliable and translatable findings; yet currently, these effects are unknown. Here, we characterized these effects for DA analyses of both relative and absolute abundances, using a combination of mathematical theory and data analysis of real and simulated case studies. We found that, for analyses based on species proportions, taxonomic bias can cause significant errors in DA results if the average measurement efficiency of the community is associated with the condition of interest. These errors can be avoided by using more robust DA methods (based on species ratios) or quantified and corrected using appropriate controls. Wide adoption of our recommendations can improve the reproducibility, interpretability, and translatability of microbiome DA studies.
Ultra-accurate microbial amplicon sequencing with synthetic long reads
Microbiome, 2021.

Background: Out of the many pathogenic bacterial species that are known, only a fraction are readily identifiable directly from a complex microbial community using standard next generation DNA sequencing. Long-read sequencing offers the potential to identify a wider range of species and to differentiate between strains within a species, but attaining sufficient accuracy in complex metagenomes remains a challenge.
Methods: Here, we describe and analytically validate LoopSeq, a commercially available synthetic long-read (SLR) sequencing technology that generates highly accurate long reads from standard short reads.
Results: LoopSeq reads are sufficiently long and accurate to identify microbial genes and species directly from complex samples. LoopSeq perfectly recovered the full diversity of 16S rRNA genes from known strains in a synthetic microbial community. Full-length LoopSeq reads had a per-base error rate of 0.005%, which exceeds the accuracy reported for other long-read sequencing technologies. 18S-ITS and genomic sequencing of fungal and bacterial isolates confirmed that LoopSeq sequencing maintains that accuracy for reads up to 6 kb in length…
Pathogen resistance may be the principal evolutionary advantage provided by the microbiome
Phil Trans B, 2020.
 To survive, plants and animals must continually defend against pathogenic microbes that would invade and disrupt their tissues. Yet they do not attempt to extirpate all microbes. Instead, they tolerate and even encourage the growth of commensal microbes, which compete with pathogens for resources and via direct inhibition. We argue that hosts have evolved to cooperate with commensals in order to enhance the pathogen resistance this competition provides. We briefly describe competition between commensals and pathogens within the host, consider how natural selection might favour hosts that tilt this competition in favour of commensals, and describe examples of extant host traits that may serve this purpose…
To survive, plants and animals must continually defend against pathogenic microbes that would invade and disrupt their tissues. Yet they do not attempt to extirpate all microbes. Instead, they tolerate and even encourage the growth of commensal microbes, which compete with pathogens for resources and via direct inhibition. We argue that hosts have evolved to cooperate with commensals in order to enhance the pathogen resistance this competition provides. We briefly describe competition between commensals and pathogens within the host, consider how natural selection might favour hosts that tilt this competition in favour of commensals, and describe examples of extant host traits that may serve this purpose…
Consistent and correctable bias in metagenomic sequencing measurements
eLife, 2019.

Marker-gene and metagenomic sequencing have profoundly expanded our ability to measure biological communities. But the measurements they provide differ from the truth, often dramatically, because these experiments are biased toward detecting some taxa over others. This experimental bias makes the taxon or gene abundances measured by different protocols quantitatively incomparable and can lead to spurious biological conclusions. We propose a mathematical model for how bias distorts community measurements based on the properties of real experiments. We validate this model with 16S rRNA gene and shotgun metagenomics data from defined bacterial communities…
High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution
Nucleic Acids Research, 2019.
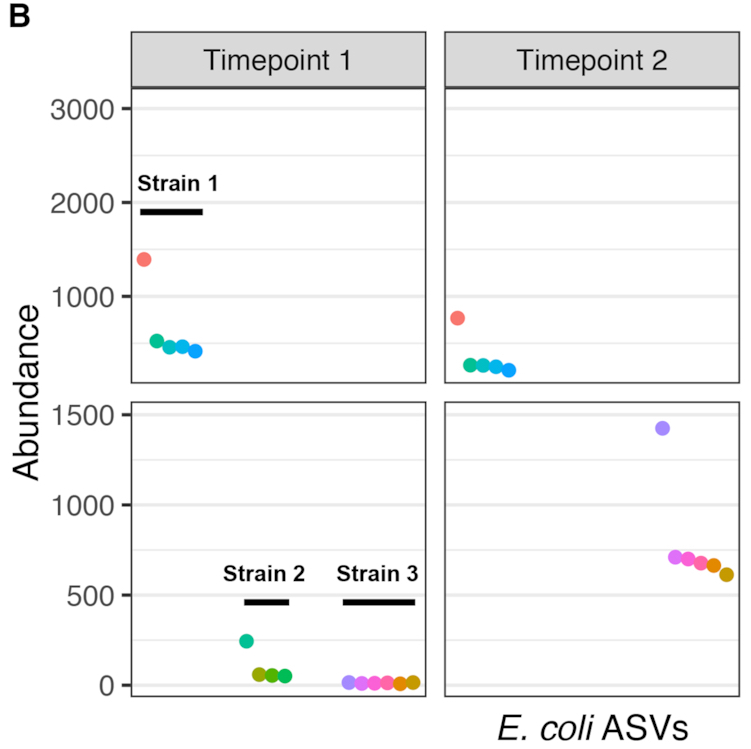
Targeted PCR amplification and high-throughput sequencing (amplicon sequencing) of 16S rRNA gene fragments is widely used to profile microbial communities. New long-read sequencing technologies can sequence the entire 16S rRNA gene, but higher error rates have limited their attractiveness when accuracy is important. Here we present a high-throughput amplicon sequencing methodology based on PacBio circular consensus sequencing and the DADA2 sample inference method that measures the full-length 16S rRNA gene with single-nucleotide resolution and a near-zero error rate. In two artificial communities of known composition, our method recovered the full complement of full-length 16S sequence variants from expected community members without residual errors….The full-length 16S gene sequences recovered by our approach allowed Escherichia coli strains to be correctly classified to the O157:H7 and K12 sub-species clades…
Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data
Microbiome, 2018.
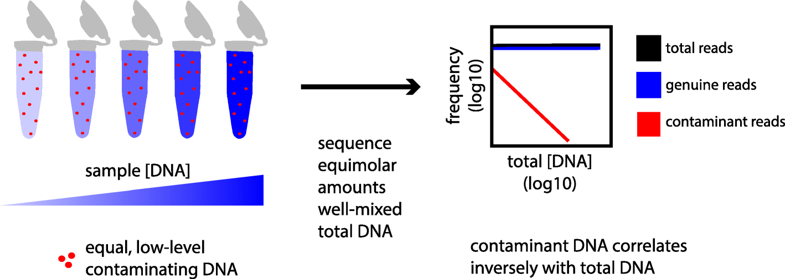
The accuracy of microbial community surveys based on marker-gene and metagenomic sequencing (MGS) suffers from the presence of contaminants—DNA sequences not truly present in the sample. Contaminants come from various sources, including reagents. Appropriate laboratory practices can reduce contamination, but do not eliminate it. Here we introduce decontam (https://github.com/benjjneb/decontam), an open-source R package that implements a statistical classification procedure that identifies contaminants in MGS data based on two widely reproduced patterns: contaminants appear at higher frequencies in low-concentration samples and are often found in negative controls…
Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women
Proceeding of the National Academy of Sciences, 2017.
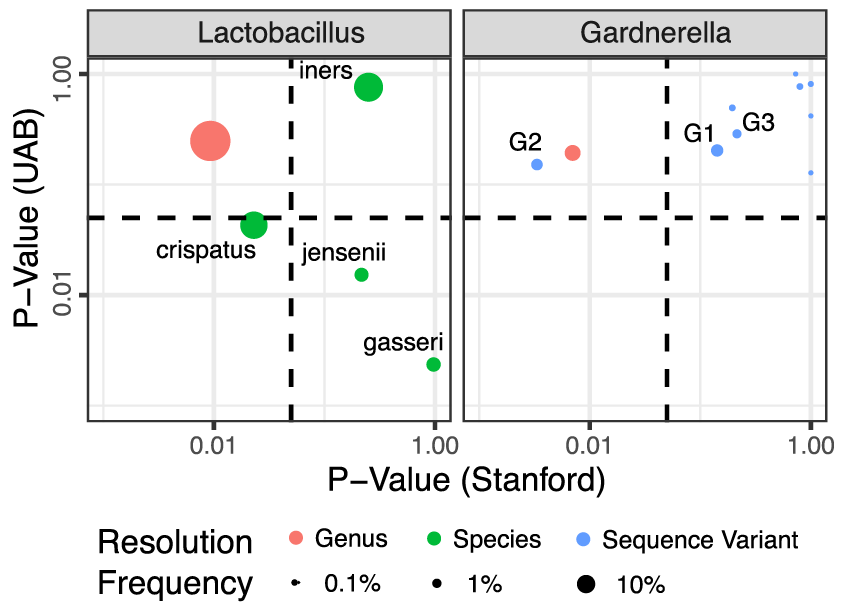
We conducted a study of PTB compared with term birth in two cohorts of pregnant women: one predominantly Caucasian (n = 39) at low risk for PTB, the second predominantly African American and at high-risk (n = 96)… Previously proposed associations between PTB and lower Lactobacillus and higher Gardnerella abundances replicated in the low-risk cohort, but not in the high-risk cohort. High-resolution bioinformatics enabled taxonomic assignment to the species and subspecies levels, revealing that Lactobacillus crispatus was associated with low risk of PTB in both cohorts, while Lactobacillus iners was not, and that a subspecies clade of Gardnerella vaginalis explained the genus association with PTB…
DADA2: high-resolution sample inference from Illumina amplicon data
Nature Methods, 2016.
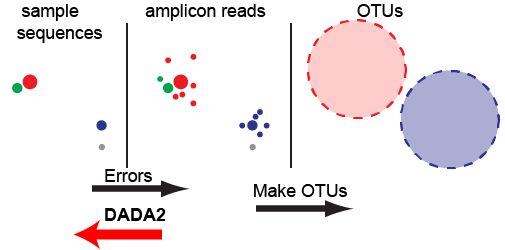
We present the open-source software package DADA2 for modeling and correcting Illumina-sequenced amplicon errors (https://github. com/benjjneb/dada2). DADA2 infers sample sequences exactly and resolves differences of as little as 1 nucleotide. In several mock communities, DADA2 identified more real variants and output fewer spurious sequences than other methods. We applied DADA2 to vaginal samples from a cohort of pregnant women, revealing a diversity of previously undetected Lactobacillus crispatus variants…
Temporal and spatial variation of the human microbiota during pregnancy
Proceeding of the National Academy of Sciences, 2015.

Despite the critical role of the human microbiota in health,our understanding of microbiota compositional dynamics during and after pregnancy is incomplete. We conducted a case-control study of 49 pregnant women, 15 of whom delivered preterm. From 40 of these women, we analyzed bacterial taxonomic composition of 3,767 specimens collected prospectively and weekly during gestation andmonthly after delivery from the vagina, distal gut, saliva, and tooth/gum…
Rapid evolution of adaptive niche construction in experimental microbial populations
Evolution, 2014
Many species engage in adaptive niche construction: modification of the local environment that increases the modifying organism’s competitive fitness. Adaptive niche construction provides an alternative pathway to higher fitness, shaping the environment rather than conforming to it… Here we report a direct observation of the de novo evolution of adaptive niche construction in populations of the bacteria Pseudomonas fluorescens… We found that adaptive niche construction emerged rapidly, within approximately 100 generations, and became ubiquitous after approximately 400 generations…
Heterozygote advantage as a natural consequence of adaptation in diploids
Proceeding of the National Academy of Sciences, 2011.
Molecular adaptation is typically assumed to proceed by sequential fixation of beneficial mutations. In diploids, this picture presupposes that for most adaptive mutations, the homozygotes have a higher fitness than the heterozygotes. Here, we show that contrary to this expectation, a substantial proportion of adaptive mutations should display heterozygote advantage. This feature of adaptation in diploids emerges naturally from the primary importance of the fitness of heterozygotes for the invasion of new adaptive mutations. We formalize this result in the framework of Fisher’s influential geometric model of adaptation…
Correlated evolution of nearby residues in Drosophilid proteins
PLoS Genetics, 2011.
Here we investigate the correlations between coding sequence substitutions as a function of their separation along the protein sequence… We find that amino acid substitutions are “clustered” along the protein sequence, that is, the frequency of additional substitutions is strongly enhanced within ≈10 residues of a first such substitution. No such clustering is observed for synonymous substitutions, supporting a “correlation length” associated with selection on proteins as the causative mechanism. Clustering is stronger between substitutions that arose in the same lineage than it is between substitutions that arose in different lineages. We consider several possible origins of clustering, concluding that epistasis (interactions between amino acids within a protein that affect function) and positional heterogeneity in the strength of purifying selection are primarily responsible…